Abstract
Inadequate light penetration in tissues restricts photodynamic therapy to treating only superficial tumors. To enable x-ray–excited photodynamic therapy (XPDT) that targets deep-seated tumors, we synthesized a nanoscintillator-photosensitizer complex containing 5% Eu-doped Y2O3 fluorescing at 611 nanometers and decorated with SiO2 containing the scintillation-coupled photosensitizer methylene blue and a polyethylene glycol coating [PEGylated Y2O3:Eu@SiO2-methylene blue (pYSM)]. When irradiated, pYSMs generate singlet oxygen species in vitro, causing cytotoxicity with hallmarks of immunogenic cell death (calreticulin translocation to the cell membrane). Intravenously administered pYSMs home passively to pancreatic tumor xenografts and, upon 10 gray irradiation, cause significant tumor regression (P < 0.01). On combining XPDT with anti-PD1 immunotherapy, a distant nonirradiated tumor also regresses via an increase in intratumoral activated CD8+ cytotoxic T cells. Collectively, we advance a systemically delivered XPDT strategy that mediates an antitumor effect in both irradiated and nonirradiated (abscopal) tumors when coupled with immunotherapy, converting an immunologically “cold” tumor to an immunologically “hot” tumor.
INTRODUCTION
Photoacoustic imaging (1), photothermal therapy (2, 3), and photodynamic therapy (4) are increasingly deployed for the diagnosis and treatment of superficial or endoscopically accessible tissues (5). However, the inability of light to traverse beyond 1 cm of tissue thickness remains a major challenge for the clinical deployment of these modalities in the diagnosis and treatment of tumors in deep-seated tissues (6). X-ray–excited photodynamic therapy (XPDT) is a recently proposed modality for treating deep tissue tumors (7). In XPDT, a scintillating nanoparticle is conjugated to a photosensitizing compound, and under x-ray excitation, the nanoscintillator emits light, which is then absorbed by the photosensitizer to generate reactive oxygen species (ROS) (8). These ROS can then induce cell death via oxidative stress through DNA or lipid membrane damage (Fig. 1Opens in image viewer). Because of the penetration depth and spatial localization of ionizing radiation systems, XPDT has the potential to supplement radiotherapy (RT) and decrease the radiation dose given to patients while improving the cytotoxic efficacy of RT (9). A critical factor in the performance of XPDT is the efficiency in ROS generation per unit dose (10). Thus, scintillators with a high light yield and photosensitizers with a high singlet oxygen quantum yield are required to produce ROS for a minimal given radiation dose.
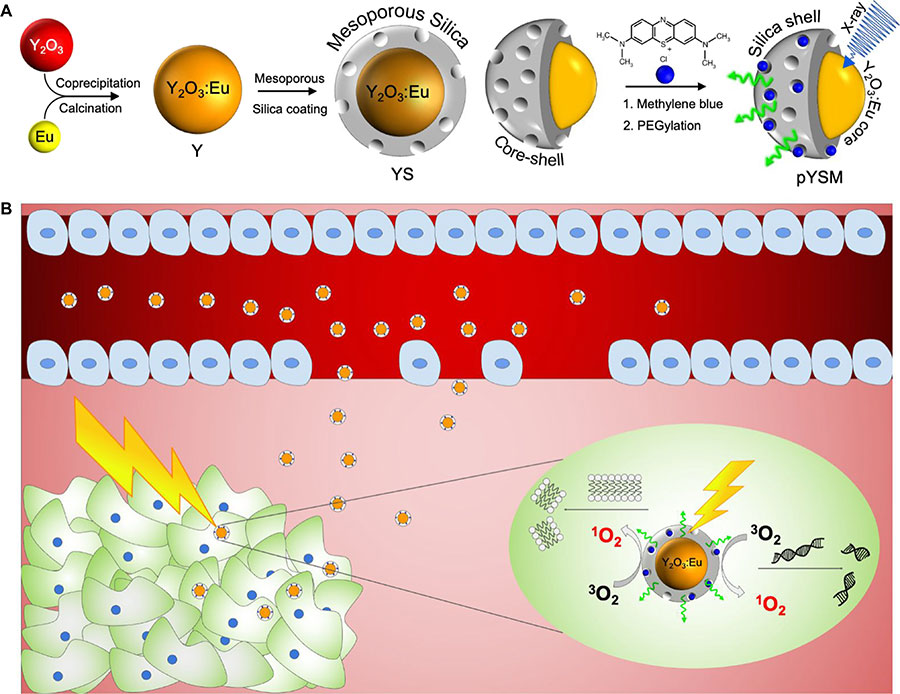
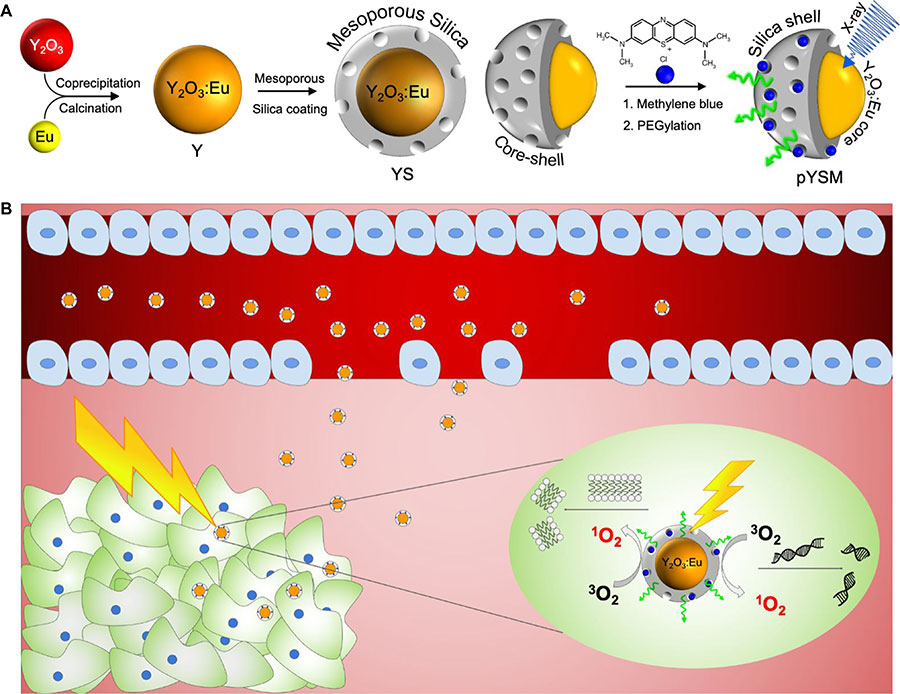
In this study, we synthesized a novel XPDT agent, PEGylated Y2O3:Eu@SiO2-methylene blue (pYSM), and demonstrated that XPDT that was performed with pYSM in combination with immune checkpoint blockade promoted a systemic antitumor immune response against solid tumors (Fig. 1, A and BOpens in image viewer). We chose Y2O3 doped with europium as a prototype phosphor because of its high light yield under x-ray excitation (11), relative ease of nanoscale fabrication, remarkable chemical inertness, and the accepted clinical use of yttria-based compounds (12). The photosensitizer was loaded into a mesoporous silica shell to couple the scintillator emission and photosensitizer absorption, this shell was then coated with polyethylene glycol (PEG) chains to increase the circulation time and biodistribution of the agent in the tumor microenvironment (13). In addition, whereas prior reports of XPDT agents have used long exposure times with low-energy x-rays [50 kilovolt peaks (kVp)], which do not penetrate well into deep tissues, our results use intravenously administered nanoparticles that accumulate in the tumor and are triggered by clinically relevant, deep-penetrating orthovoltage x-rays (320 kVp) (fig. S1) and define a heretofore unexplored synergy with immunotherapy. An abscopal effect was observed in mice with bilateral tumors where right-sided tumors treated with combination XPDT and immunotherapy also resulted in a significant size reduction of untreated left-sided tumors. To our knowledge, this is the first work assessing the systemic immune response of XPDT combined with immunotherapy. The results found in this study suggest that XPDT in combination with checkpoint blockade could change the immune profile of solid tumors and increase their sensitivity to a systemic antitumor immune response.
RESULTS
XPDT agent synthesis
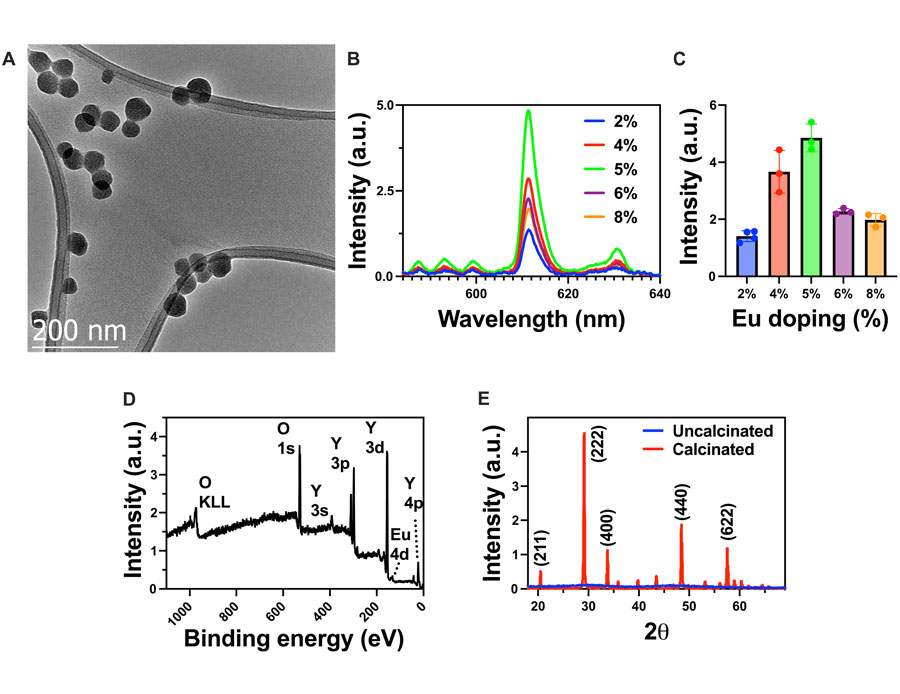
Scintillating Y2O3:Eu nanoparticles were synthesized via an aqueous coprecipitation method. YCl3 and EuCl3 were mixed in deionized (DI) water with urea as a precipitating agent and 50-kDa polyvinyl pyrrolidone as a capping agent to control the size and polydispersity of the nanoparticles (14, 15). The resulting rare earth hydroxycarbonates were calcined in open air to yield the scintillating binary oxide. A custom-built optical setup was used to measure the light output of the nanoscintillators excited by 320-kVp x-rays (figs. S2 and S3). To optimize the light yield of the scintillators, the europium doping concentration was optimized. As seen in Fig. 2AOpens in image viewer, transmission electron microscopy (TEM) images show that the resulting binary oxide nanoparticles are well segregated and have a uniform diameter of ~50 nm. High-resolution TEM also showed a characteristic plane along the [101] zone axis because of the typical orientation of planes (200) and (220) with d-spacings of 0.51 and 0.8 nm, respectively (fig. S4). Figure 2BOpens in image viewer shows the radioluminescence spectra of the Y2O3:Eu nanoparticles under x-ray excitation. The peak near 611 nm is due to the 5D0 → 7F2 transition, which is characteristic of the europium dopant and remains consistent across different doping concentrations (11). Figure 2COpens in image viewer shows the relationship between the doping concentration of europium and the resulting light yield from 2 to 8% europium doping. When the doping concentration is low, adding more dopant ions increases the light yield. Beyond 5% doping, the light yield begins to decrease as dopant ions are added, which can be due to a self-quenching process where the europium luminescence centers absorb each others’ light emission, resulting in an overall decrease in light yield (16). X-ray photoelectron spectroscopy (XPS) and x-ray diffraction (XRD) were performed (Fig. 2, D and EOpens in image viewer) to further confirm the presence of europium and the crystallinity of the yttria lattice after calcination (fig. S5) (17). XRD peaks matched cubic phase Y2O3 (JCPDS #71-0099).
In vitro XPDT studies
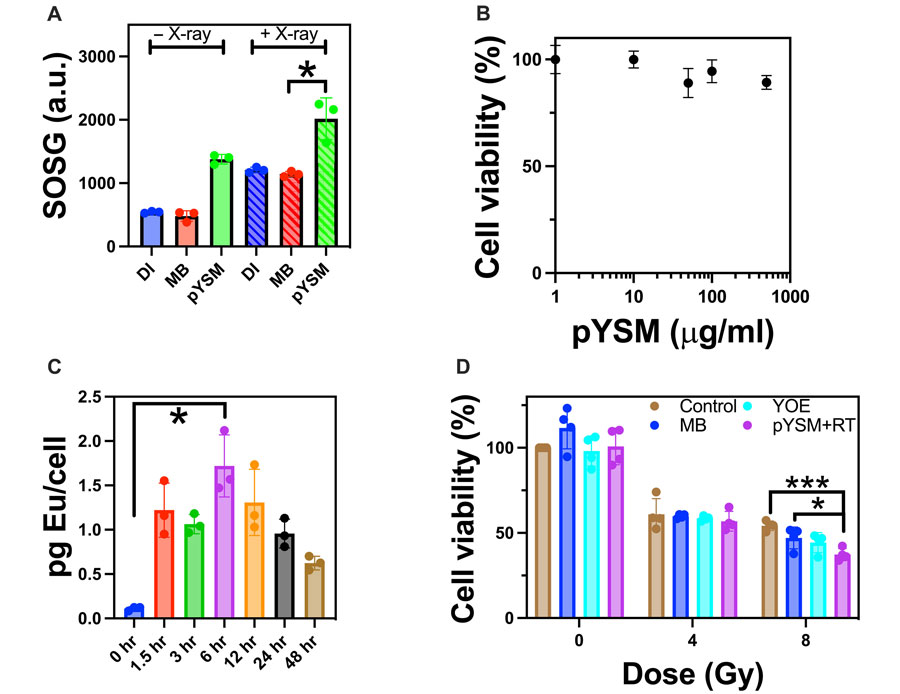
To characterize x-ray–excited ROS generation, a singlet oxygen sensor green (SOSG) assay was performed with pYSM compared to methylene blue and DI water as controls (20). As seen in Fig. 4AOpens in image viewer, the pYSM group produced the largest statistically significant amount of singlet oxygen upon irradiation compared to controls, showing that Y2O3:Eu is required to activate methylene blue upon x-ray excitation. Notably, there was no appreciable increase in temperature ruling out a photothermal mechanism of action (fig. S8). Encouraged by these results, we incubated the pYSM agent at varying concentrations with murine Panc02 pancreatic cancer cells to measure the toxicity of the XPDT agent. As seen in Fig. 4BOpens in image viewer, cells remained viable at concentrations above 100 μg/ml. In addition, cellular uptake studies were performed via inductively coupled plasma mass spectrometry (ICP-MS), which showed that 6 hours of incubation resulted in the maximal pYSM concentration within cells (Fig. 4COpens in image viewer). Analysis of variance followed by Tukey’s post hoc test was performed, showing a statistically significant difference in cellular uptake between 0 and 6 hours of incubation. To test the in vitro consequences of XPDT-mediated singlet oxygen production, we treated Panc02 cells with pYSM particles and respective controls and split them into irradiated and nonirradiated groups (Fig. 4DOpens in image viewer). While no noticeable cell death occurred in the nonirradiated group, cell viability decreased significantly after a dose of 8–gray (Gy) irradiation in cells cultured with pYSM when compared to the control and pYSM-only groups, suggesting that pYSM could be used as an XPDT agent.
In vivo XPDT studies
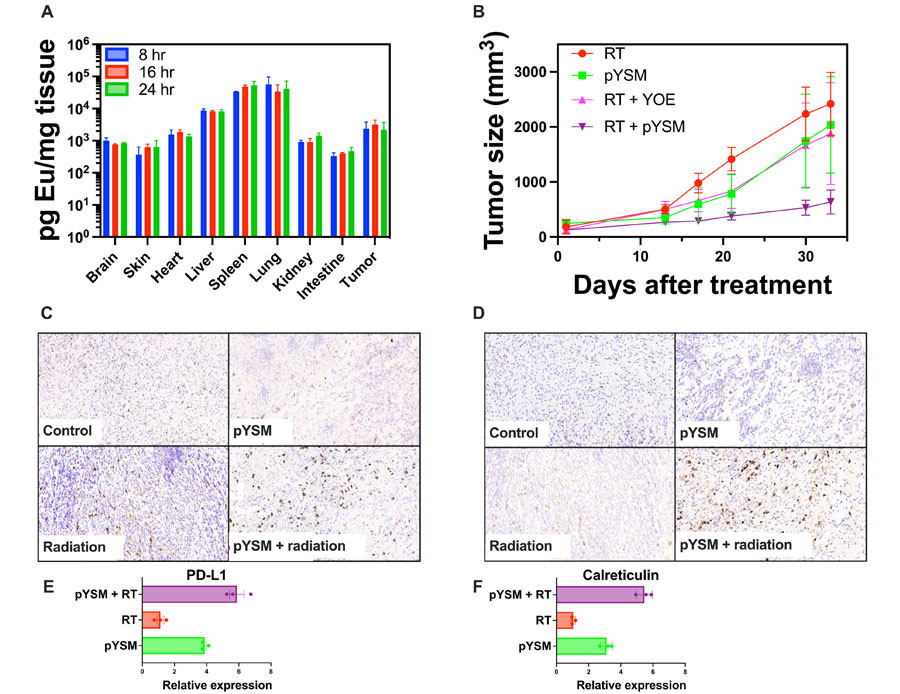
To be effective as an XPDT agent, systemically delivered pYSM must accumulate in the target tumor. In a biodistribution analysis conducted in mice with subcutaneous Panc02 tumors, normal organs and tumors were harvested at 8, 16, and 24 hours following the intravenous administration of pYSM. Tumor uptake was noted to be relatively constant at all time points with no statistically significant difference in uptake, while the liver, spleen, and lungs showed high uptake as well (Fig. 5AOpens in image viewer). On the basis of this, we proceeded to perform a tumor regrowth delay experiment with pYSM administered intravenously 24 hours before irradiation with a single dose of 10 Gy. A statistically significant decrease in tumor volume was noted in the pYSM + radiation group compared to radiation alone (Fig. 5BOpens in image viewer). Immunohistochemical analysis of the tumors harvested from these mice showed an increase in the CD8+ and CD4+ T cells in the XPDT-treated tumors (Fig. 5, C and DOpens in image viewer). Surprised by this finding, we went back to test the in vitro immunogenicity of XPDT and observed that XPDT increased the expression of both calreticulin and programmed death ligand 1 (PD-L1) on the surface of Panc02 cells when compared to just RT or only particle incubation. This indicated that not only was there an immunogenic cell death response (21), but there was also an immunosuppressive (22), yet druggable, phenotype activated as well (Fig. 5, E and FOpens in image viewer). This experiment suggested that the antitumor activity of XPDT therapy could be enhanced if coupled with immunotherapy aimed at boosting the immune response mounted against the tumor. Mouse weights were also separately tracked for the control, RT-only, pYSM-only, and XPDT groups and showed no appreciable difference across the treatment groups, suggesting no overt toxicity of the pYSMs (fig. S9).
XPDT combined with immunotherapy
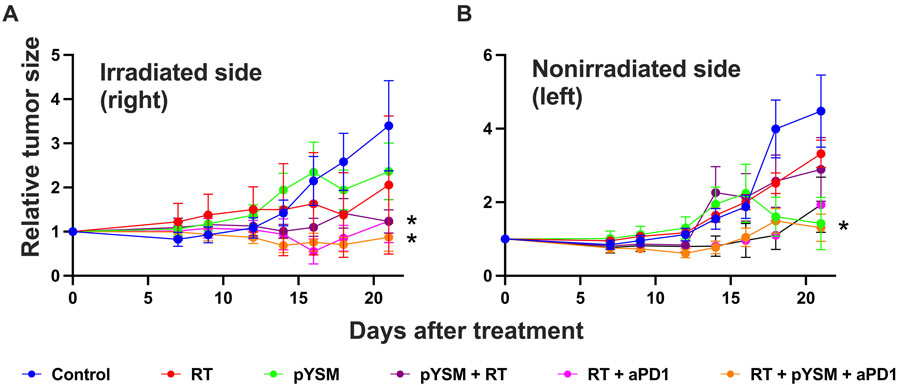
To test this hypothesis, ovalbumin-expressing Panc02 (Panc02-Ova) cells were subcutaneously implanted in both thighs of immune-competent C57BL/6 mice and treated with a combination of XPDT and anti-PD1 immunotherapy. The tumors on the right thigh were treated, but the tumors on the left thigh were not. These nonirradiated tumors served as surrogates for distant or metastatic disease, allowing us to interrogate abscopal responses driven by the activation of a robust systemic immune response. Our analysis of the right-sided tumor showed that pYSM + RT significantly decreased the tumor growth, as previously demonstrated (Fig. 5BOpens in image viewer), but also that this tumor growth inhibition was significantly augmented by the addition of anti-PD1 therapy (Fig. 6AOpens in image viewer). Compared to the control group, two groups of mice had statistically significant regression in tumor size: mice given either XPDT or XPDT + anti-PD1 therapy. Similarly, the left-sided tumors in the XPDT + anti-PD1 group had a statistically significant decrease in tumor volume, suggesting that a robust systemic immune response was activated (Fig. 6BOpens in image viewer).
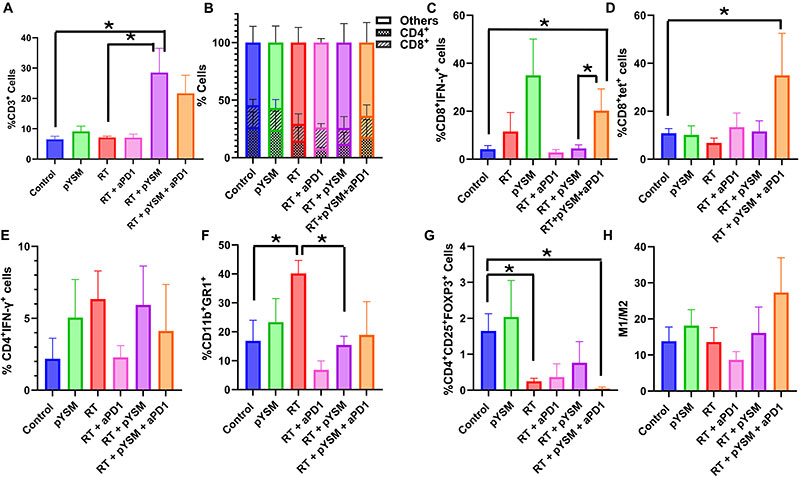
To further characterize this effect, we performed immunophenotyping of tumor-infiltrating lymphocytes harvested from both tumors using flow cytometry. In the treated right-sided tumors, XPDT therapy significantly increased T cell infiltration in the tumor, as indicated by the significant increase in CD3+ T cells (inclusive of CD4+ and CD8+ T cells) in the RT + pYSM–treated tumors when compared to the control group (Fig. 7, A and BOpens in image viewer). While the addition of anti-PD1 therapy to RT + pYSM did not significantly increase the infiltration of tumors by T cells, it increased the proportion of the activated CD8+IFN-γ+ T cells and elicited a trend toward increased antigen-specific CD8+tet+ T cells (Fig. 7, C and DOpens in image viewer). Analysis of the immunosuppressive cell populations showed that radiation by itself and RT + pYSM + aPD1 decreased the number of CD4+CD25+FOXP3+ regulatory T cells (Tregs) when compared to untreated control tumors (Fig. 7GOpens in image viewer). While there were modest, nonstatistically significant differences in the proportion of interferon-γ (IFN-γ)–secreting CD4+ T cells [T helper 1 (TH1) cells] between the groups (Fig. 7EOpens in image viewer), there was a significant decrease in the CD11b+GR1+ myeloid-derived suppressor T cell (MDSC) population in between the RT-only group versus the control and RT + pYSM groups (Fig. 7FOpens in image viewer). While anti-PD1 therapy slightly increased the M1/M2 macrophage ratio in treated tumors, no statistically significant response was observed (Fig. 7HOpens in image viewer).
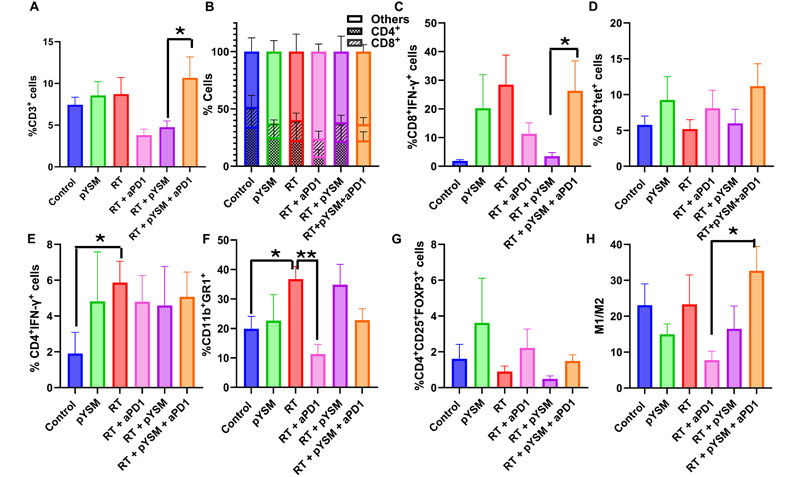
Analysis of the left tumors, which were not irradiated, showed that neither the RT group nor the RT + pYSM group showed an increase in the CD3+ population (Fig. 8AOpens in image viewer). However, anti-PD1 in combination with radiation and pYSM significantly increased the infiltration of CD3+ T cells (inclusive of CD4+ and CD8+ T cells) in the nonirradiated tumors compared to nonirradiated tumors in the pYSM + RT group (Fig. 8, A and BOpens in image viewer). Anti-PD1 therapy in combination with RT and pYSM significantly increased the activated CD8+IFN-γ+ T cells when compared to the RT + pYSM–treated counterparts (Fig. 8COpens in image viewer). However, there was no statistically significant increase in antigen-specific T cells in the left tumors of mice between the different treatment groups (Fig. 8DOpens in image viewer). None of the treatment groups significantly altered the Treg population in the nonirradiated tumors (Fig. 8GOpens in image viewer). Similar to the irradiated right tumor, RT significantly increased the population of CD11b+GR1+ MDSCs, but unlike the irradiated tumors, an increase in IFN-γ–secreting TH1 cells was also noted in the RT group in the tumor microenvironment (Fig. 8, E and FOpens in image viewer). When comparing the RT alone and RT + aPD1 groups, the addition of anti-PD1 therapy decreased MDSCs in nonirradiated (left) tumors (Fig. 8FOpens in image viewer). However, none of the other treatment groups significantly altered the MDSC population in the tumor microenvironment of the nontreated tumor compared to the control (Fig. 8FOpens in image viewer). Notably, the M1/M2 macrophage ratio in the left tumor was significantly higher in the RT + pYSM + aPD1 group compared to the RT + aPD1 group (Fig. 8GOpens in image viewer). Collectively, these results suggest a shift in the tumor immune microenvironment favoring a pro-inflammatory antitumor phenotype when XPDT is coupled with immunotherapy.